


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
新井 栄揮; 玉田 太郎; 前田 宜丈*; 黒木 良太
no journal, ,
マウス抗体TN1は、巨核球系細胞の増殖・分化及び血小板産生を促進するサイトカインであるヒト・トロンボポエチン(hTPO)を認識する。TN1抗体のTPO中和活性は、TPOによるTPO受容体の二量体化が阻害されるからであると考えられている。本研究では、抗原との結合によるTN1の構造変化を調べるために、TN1由来FabのX線結晶構造解析を2.1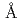 分解能で決定し、TN1-Fab/hTPO複合体(PDB id 1V7M)のFabと比較を行った。その結果、TN1由来Fabを構成している各構造ドメインは、抗原認識部位である超可変領域(CDR領域)を含め、抗原結合時・非結合時においてほとんど変化が見られなかった(rms deviation
分解能で決定し、TN1-Fab/hTPO複合体(PDB id 1V7M)のFabと比較を行った。その結果、TN1由来Fabを構成している各構造ドメインは、抗原認識部位である超可変領域(CDR領域)を含め、抗原結合時・非結合時においてほとんど変化が見られなかった(rms deviation 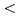 0.6)。抗原結合時と比較して、非結合時は可変領域・一定構造領域の相対位置がわずかにずれるが、これは結晶中の分子のパッキングの差によるものと思われる。これらの結果は、TN1由来Fabが抗原との結合の際に構造変化を必要としないことを示唆する。
0.6)。抗原結合時と比較して、非結合時は可変領域・一定構造領域の相対位置がわずかにずれるが、これは結晶中の分子のパッキングの差によるものと思われる。これらの結果は、TN1由来Fabが抗原との結合の際に構造変化を必要としないことを示唆する。
石田 恒; 郷 信広
no journal, ,
ホリデイ構造は、DNA相同組換えの普遍的なDNA中間体である。RuvAはホリデイ構造特異的DNA結合蛋白質である。RuvBはホリデイ分岐DNAと結合し、ATP加水分解エネルギーを用いてホリデイ分岐点の移動反応を触媒する。昨年度は、E.Coli由来のRuvA4量体-ホリデイ分岐DNA複合体のアンブレラサンプリングシミュレーションを用いて、分岐点移動におけるエネルギー地形を解析し、ホリデイ分岐DNAの分岐点移動の仕組みを原子レベルで説明した。しかしながら、このシミュレーションではRuvBは陽には含まれていなかったため、RuvBのダイナミクスを解析することはできなかった。本年度は、さらにRuvBの構造も入れたRuvA-RuvB-ホリデイ分岐DNA複合体の分子動力学シミュレーションを実行した。この複合体はX線結晶解析では解かれていないため、部分的に解かれている部分構造を用いて全体構造をモデル化した。モデル化された系の原子数は約50万個となった。この系の分子動力学シミュレーションを5ナノ秒間実行した。そして主成分解析を用いてホリデイ構造モデルのダイナミクスを解析した。結果、RuvBのダイナミクスが分岐点移動と関係していることがわかった。
中川 洋; 城地 保昌*; 北尾 彰朗*; 柴田 薫; 郷 信広; 片岡 幹雄
no journal, ,
タンパク質のボソンピークや動力学転移の水和の効果を中性子非弾性散乱により調べた。極低温では3 4meVにボソンピークが観測され、ピーク位置は水和により高エネルギー側へシフトすることがわかった。ボソンピーク近傍のスペクトルが示すタンパク質の低振動モードは調和振動的であり、ピークのシフトからそのばね定数は水和量が多いほど大きくなると言える。これは水素結合を介した水和水とタンパク質の相互作用によってタンパク質の低振動モードのエネルギー地形がより凸凹になったことに起因し、このことはシミュレーションからの理論的な予測(Y. Joti et al.,2005)と一致する。一方、水和量が約0.2(g water/g protein)以上で240K付近において動力学転移が観測された。なぜ動力学転移が水和依存的に生じるのかを調べるために、中性子散乱の同位体効果を利用して水和水のダイナミクスを直接観測した。その結果、転移温度以下の低温では水和量に関係なくタンパク質と水分子の揺らぎの大きさはほぼ同じであった。また転移が生じない低い水和量の場合では転移温度以上でもやはりタンパク質とほぼ同じであった。一方、動力学転移が生じる時には同時に水和水の揺らぎが大きくなっていることが明らかになった。高い水和量で生じるタンパク質表面の水分子の特異的なダイナミクスが、タンパク質と水分子の界面に存在する水素結合ネットワークを介してタンパク質の振動モードと相互作用し、その結果動力学転移が生じると考えている。
4meVにボソンピークが観測され、ピーク位置は水和により高エネルギー側へシフトすることがわかった。ボソンピーク近傍のスペクトルが示すタンパク質の低振動モードは調和振動的であり、ピークのシフトからそのばね定数は水和量が多いほど大きくなると言える。これは水素結合を介した水和水とタンパク質の相互作用によってタンパク質の低振動モードのエネルギー地形がより凸凹になったことに起因し、このことはシミュレーションからの理論的な予測(Y. Joti et al.,2005)と一致する。一方、水和量が約0.2(g water/g protein)以上で240K付近において動力学転移が観測された。なぜ動力学転移が水和依存的に生じるのかを調べるために、中性子散乱の同位体効果を利用して水和水のダイナミクスを直接観測した。その結果、転移温度以下の低温では水和量に関係なくタンパク質と水分子の揺らぎの大きさはほぼ同じであった。また転移が生じない低い水和量の場合では転移温度以上でもやはりタンパク質とほぼ同じであった。一方、動力学転移が生じる時には同時に水和水の揺らぎが大きくなっていることが明らかになった。高い水和量で生じるタンパク質表面の水分子の特異的なダイナミクスが、タンパク質と水分子の界面に存在する水素結合ネットワークを介してタンパク質の振動モードと相互作用し、その結果動力学転移が生じると考えている。
堤 遊*; 松本 淳; 由良 敬; 石田 恒
no journal, ,
近年、X線結晶解析により高分解能で解かれた生体超分子を構成する要素分子(単体の蛋白質,核酸など)を電子顕微鏡による低分解能の生体超分子像にあてはめることにより、生体超分子の立体構造モデルを構築することが可能になりつつある。本研究では、遺伝子情報翻訳装置であるリボソームの原子分解能の立体構造を、電子顕微鏡像とX線結晶解析のデータ及び分子シミュレーションの手法を用いて構築し、リボソームの機能を原子レベルで理解することを目的とする。われわれは電子顕微鏡像を考慮した拘束条件のもと分子シミュレーションを実行するプログラムを開発し、より現実的なリボソーム立体構造の精密化を実行した。リボソームの電子像データにはEMBL-EBIに登録されている遺伝子情報翻訳開始から終了までの6つのデータを用いた。そして、それぞれの反応条件下の立体構造を構築し、tRNA部位,生成ペプチド鎖のトンネル部位を詳しく解析した。結果、リボソームの機能と構造の関係を見いだすことができた。
松本 淳
no journal, ,
最新の1分子DNA計測実験により決定されたDNAのねじれに関する力学定数は、今まで考えられていた値よりもかなり大きい。この理由を明らかにするために、われわれが新たに開発した粗視化DNAモデルの基準振動解析法を改良し、DNA2重らせんのねじれ特性を研究できるようにした。われわれの計算では、実験同様にDNAの両端に外力がかけられている場合、塩基対がDNAのらせん軸に対して傾いている場合、さらに、DNAの局所構造があるパターンに従って変化する場合には、力学定数が大きくなることが示された。
 1 chain and its interaction with its ligand
1 chain and its interaction with its ligand黒木 良太; 本庄 栄二郎; 玉田 太郎; 有馬 和彦*; 出原 賢治*
no journal, ,
インターロイキン-13は、IL-13受容体a1鎖(IL-13Ra1)及びIL-4受容体a鎖(IL-4Ra)という2つの受容体と相互作用し、そのシグナルを伝達する。われわれは既にIL-13Ra1のイムノグロブリン様ドメインが、IL-13の認識に重要であることを見いだしている。その相互作用様式を立体構造的に明らかにするため、IL-13Ra1及びIL-4Raの二つの受容体の細胞外領域をコードする遺伝子を抗体のFc領域の遺伝子に融合させ、さらにカイコ蛾による発現システムを用いて生産した。発現された蛋白質をProtein Gカラムを用いて精製した後、Fc領域をプロテアーゼ消化によって除去、さらに陰イオン交換クロマトグラフィーにより精製した。こうして得られたIL-13Ra1及びIL-4RaとリガンドであるIL-13との相互作用をゲル濾過を用いて解析した。その結果、IL-13は単独でIL-4Raと強く結合するが、IL-13Ra1との相互作用は弱いことがわかった。しかしながらIL-13Ra1及びIL-4Rの両方が存在するときには、IL-13の親和性は著しく向上し、3つの分子が複合体を形成することがわかった。
樋口 真理子; Pinak, M.
no journal, ,
クラスター損傷とは1020ベースペア内に二つ以上の損傷があるものをいい、放射線により高い頻度で生じる。クラスター損傷は単独の損傷よりも修復されにくく、その修復機構もよくわかっていない。8オキソグアニン(8oxoG)の近傍に塩基欠落部位(APサイト)があるクラスター損傷は修復が困難な損傷の一つである。実験により、8oxoGのある鎖の、対の鎖上の8oxoGから1ベースペア離れた位置にAPサイトがあると、グリコシラーゼMutMによって8oxoGが修復されにくくなること、また、8oxoGとAPサイトの距離が離れると、修復される確率が増えていくことがわかっている。本研究では、単独の8oxoG損傷を持つDNAと8oxoG-APサイトによるクラスター損傷DNAの分子動力学シミュレーションを行い、結果を比較した。分子動力学シミュレーションを行うために、損傷部位の力場パラメータを量子化学計算により求めた。分子構造と原子電荷分布が損傷のないDNAとは異なることがわかった。分子動力学シミュレーションではAPサイトがクラスター損傷の形状に大きく影響していることがわかった。
藤原 悟
no journal, ,
小角散乱法は、溶液中の蛋白質等の非結晶系の構造研究に有用な方法である。特に中性子小角散乱はコントラスト変調法や選択的重水素化技術と組合せることにより、複合体中の特定成分の構造情報を得るというユニークな特徴を持つ。われわれは、中性子小角散乱法を用いて、筋収縮及び制御系に関係した蛋白質複合体の研究を行ってきた。骨格筋,心筋において筋収縮制御はトロポニン-トロポミオシン-アクチンから構成される筋肉の細いフィラメント中において行われている。トロポニンは3種類の成分(トロポニンC, I, T)から成る蛋白質複合体である。筋収縮はCa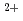 のトロポニンCへの結合によって開始される。細いフィラメント中におけるトロポニンの構造を知ることは収縮制御の分子機構を理解するうえで重要である。われわれは、重水素化トロポニンCを調製し、細いフィラメント中に挿入し、中性子小角散乱実験及び中性子繊維回折測定を行うことにより、細いフィラメント中でのトロポニンCの構造(変化)を明らかにした。本講演では、その実験についての報告を行い、中性子散乱の有用性について議論する。
のトロポニンCへの結合によって開始される。細いフィラメント中におけるトロポニンの構造を知ることは収縮制御の分子機構を理解するうえで重要である。われわれは、重水素化トロポニンCを調製し、細いフィラメント中に挿入し、中性子小角散乱実験及び中性子繊維回折測定を行うことにより、細いフィラメント中でのトロポニンCの構造(変化)を明らかにした。本講演では、その実験についての報告を行い、中性子散乱の有用性について議論する。
河野 秀俊; 皿井 明倫*
no journal, ,
遺伝子発現は、転写因子によって制御されている。転写因子は、DNA配列に特異的に結合するが、似たような一群の配列に結合する。また、転写因子は他の転写因子と協同的に結合し、その配列特異性に多様性をもたらす。そのような多様性がどのように生じるのか、構造バイオインフォマティクスのアプローチにより解析を行った。
Kim, O.*; 由良 敬; 郷 信広
no journal, ,
タンパク質とRNAの相互作用は重要な働きを担っている。しかしその複合体構造はよくわかっていない。そこで、わずかにわかっているタンパク質とRNAの複合体構造を元にコンピュータ統計解析を行った。その結果、タンパク質のどのような部分にRNAは相互作用するかを見いだすことに成功し、さらにその統計量を用いてタンパク質の立体構造からRNA相互作用面を高い精度で推定することを可能にした。なお、この手法をmRNA輸送システムに適用することも試みている。
吉田 智喜*; 土井 英雄*; 相田 美砂子*; 河野 秀俊; Kumar, S.*; Gromiha, M. M.*; 皿井 明倫*
no journal, ,
蛋白質がどのように配列特異的にDNAに結合するか明らかにするために、20種類のアミノ酸残基と塩基対において、相互作用の自由エネルギー地形を計算した。配列を変えたオリゴDNAをある座標系に固定し、アミノ酸残基をその周囲に配置させ、相互作用エネルギーを計算した。マルチカノニカルモンテカルロ法でアミノ酸残基の位置及び構造を効率的に変えながら、エンタルピー,エントロピー,自由エネルギー地形を描いた。得られた地形は、蛋白質-DNAの複合体構造に見られる相互作用をよく再現した。
米谷 佳晃*; 河野 秀俊; 郷 信広
no journal, ,
DNA構造は配列依存性がある。これまで、2塩基対の配列(10通り)の構造変形について調べられてきたが、その前後の塩基対の構造への影響も大きいことが指摘されている。そこで、4塩基対(136通り)の分子動力学計算を行い、構造変形のしやすさを調べた。その結果、構造変形のしやすさとDNAの水和が極めて高い相関があることがわかった。
藤井 聡*; 河野 秀俊; 竹中 繁織*; 郷 信広; 皿井 明倫*
no journal, ,
DNAの分子動力学計算を行い、リン酸骨格の構造に注目して解析を行った。リン酸骨格はA型,B型DNA構造を決める指標になっており、蛋白質が結合した構造ではA型構造に構造が変化するDNA配列が知られている。そこで、その構造転移が配列固有の特性によるものかどうか調べるために、DNA配列とリン酸骨格の構造の関係を解析した。その結果、構造転移を起こしやすい配列が存在することがわかった。その配列は既に知られているA型に構造転移する配列をも含んでいた。これらの結果は、DNAの物性自体も蛋白質との相互作用に対する情報を持つことを示す。
藤井 聡*; 河野 秀俊; 竹中 繁織*; 郷 信広; 皿井 明倫*
no journal, ,
DNA構造は配列によって構造変形のしやすさが異なることが知られている。近年、われわれはDNAの曲がりやすさが蛋白質のDNA認識において重要な要因であることを指摘している。本研究では、DNA配列のまがりやすさを定量化するために、さまざまなDNA配列の分子動力学計算を行った。サンプリングした構造アンサンブルからDNAの構造変形のしやすさを計算したところ、プリン-ピリミジンの並びは前後の塩基対にかかわらず似たような構造をとる、つまり、硬い構造であることがわかった。一方、ピリミジン-プリンの並びでは前後の塩基対に構造が大きく依存していることがわかった。蛋白質が結合する配列は柔らかい配列が多い傾向が見られた。
Ngahu, A.*; 上野 卓哉*; Ahmad, S.*; 河野 秀俊; 皿井 明倫*
no journal, ,
酵母ゲノムにおいて、DNA結合蛋白質を推定し、その推定したDNA結合蛋白質のターゲット配列をさらに推定した。これにより、どの遺伝子がどの遺伝子と関連があるか予測することができる。酵母の場合、約150のDNA結合蛋白質が推定された。同じターゲット配列を持つ遺伝子は、細胞周期の同時期に働くと考えられており、その産物である蛋白質は、お互いに相互作用する可能性が高い。これをもとに、蛋白質-蛋白質相互作用の推定に応用できる。